
Ubiquitin signalling.

Proteins are targeted for degradation by the proteasome with covalent modification of a lysine residue that requires the coordinated reactions of three enzymes. In the first step, a ubiquitin-activating enzyme (known as E1) hydrolyzes ATP and adenylylates a ubiquitin molecule. This is then transferred to E1's active-site cysteine residue in concert with the adenylylation of a second ubiquitin. This adenylylated ubiquitin is then transferred to a cysteine of a second enzyme, ubiquitin-conjugating enzyme (E2). In the last step, a member of a highly diverse class of enzymes known as ubiquitin ligases (E3) recognizes the specific protein to be ubiquitinated and catalyzes the transfer of ubiquitin from E2 to this target protein. A target protein must be labeled with at least four ubiquitin monomers (in the form of a polyubiquitin chain) before it is recognized by the proteasome lid.[32] It is therefore the E3 that confers substrate specificity to this system.[33] The number of E1, E2, and E3 proteins expressed depends on the organism and cell type, but there are many different E3 enzymes present in humans, indicating that there is a huge number of targets for the ubiquitin proteasome system. The mechanism by which a polyubiquitinated protein is targeted to the proteasome is not fully understood. Ubiquitin-receptor proteins have an N-terminal ubiquitin-like (UBL) domain and one or more ubiquitin-associated (UBA) domains. The UBL domains are recognized by the 19S proteasome caps and the UBA domains bind ubiquitin via three-helix bundles. These receptor proteins may escort polyubiquitinated proteins to the proteasome, though the specifics of this interaction and its regulation are unclear.[34] The ubiquitin protein itself is 76 amino acids long and was named due to its ubiquitous nature, as it has a highly conserved sequence and is found in all known eukaryotic organisms. The genes encoding ubiquitin in eukaryotes are arranged in tandem repeats, possibly due to the heavy transcription demands on these genes to produce enough ubiquitin for the cell. It has been proposed that ubiquitin is the slowest-evolving protein identified to date.
SMRT DNA Sequencing

Pacific biosciences is developing a transformed DNA sequencing technology, which will revolutionize the field of genetic analysis by enabling researchers to answer questions important to human healthcare, It is called SMRT (Single Molecule Real-Time DNA Sequencing). A breakthrough technology based on the natural process that occurs every time living cells divides.

 Subscribe in a reader
Subscribe in a reader Prior to division DNA is replicated by enzymes called DNA polymerase is which efficiently duplicating entire genomes in minutes, by reading the DNA and sequentially building a complementary strand with matching building blocks called nucleotides. Pacific biosciences SMRT sequencing harnesses the power of the polymerase as sequencing engine by eavesdropping on what works to replicate DNA, this approach is enabled by two proprietary technologies the first Phospholinked nucleotides. To visualize polymerase activity a different colored fluorescent label is attached to each of the four nucleotides ACG and T. In contrast to other sequencing approaches R phosholinked nucleotides carry their fluorescent label on the terminal phosphate rather than the base. Through this innovation enzyme cleaves away the fluorescent label as part of the incorporation process leaving behind a completely natural strand of DNA.This enables us to exploit the inherent properties of the DNA polymerase including high-speed long read length and high fidelity. The second key technology is a nano photonic visualization chamber called the Zero Mode Wave-guide or ZMW, It enables observation of the individual molecules against a required background of labeled nucleotides while maintaining a high signal-to-noise. This ZNW is a cylindrical metallic chamber approximately 70 nm wide it is illuminated/support creating an extremely small detection volume just 20 cL. Nucleotides defused in and out of the ZNW in microsecond. When the polymerase encounters the correct nucleotide it takes several milliseconds to incorporated during which time its florescent label is excited emitting light is captured by a sensitive detector. After incorporation the label is clipped off and diffuses away, The whole process repeats creating sequential bursts of light corresponding to the different nucleotides these are recorded thus building the DNA sequence.
Application
The Single Molecule Real Time sequencing will be applicable for a broad range of genomics research, namely:- De novo genome sequencing: The read length from the Single Molecule Real Time sequencing is currently comparable to that from the Sanger sequencing method based on dideoxynucleotide chain termination. The longer read length allows de novo genome sequencing and easier genome assemblies.
- Individual whole genome sequencing: Individual genome sequencing may utilize the Single Molecule Real Time sequencing method for the personalized medicine.
- Resequencing: A same DNA molecule can be resequenced independently by creating the circular DNA template and utilizing a strand displacing enzyme that separates the newly synthesized DNA strand from the template.


Effux pumps

Active efflux is a mechanism responsible for extrusion of toxic substances and antibiotics outside the cell; this is considered to be a vital part of xenobiotic metabolism. This mechanism is important in medicine as it can contribute to bacterial antibiotic resistance.
Efflux systems function via an energy-dependent mechanism (Active transport) to pump out unwanted toxic substances through specific efflux pumps. Some efflux systems are drug-specific, whereas others may accommodate multiple drugs, and thus contribute to bacterial multidrug resistance (MDR).
Bacterial efflux pumps
Efflux pumps are proteinaceous transporters localized in the cytoplasmic membrane of all kinds of cells. They are active transporters, meaning that they require a source of chemical energy to perform their function. Some are primary active transporters utilizing Adenosine triphosphate hydrolysis as a source of energy, whereas others are secondary active transporters (uniporters, symporters, or antiporters) in which transport is coupled to an electrochemical potential difference created by pumping out hydrogen or sodium ions outside the cell.
Bacterial efflux transporters are classified into five major superfamilies, based on the amino acid sequence and the energy source used to export their substrates:1. The major facilitator superfamily (MFS)
2. The ATP-binding cassette superfamily (ABC)
3. The small multidrug resistance family (SMR)
4. The resistance-nodulation-cell division superfamily (RND)
5. The Multi antimicrobial extrusion protein family (MATE).
Of these, only the ABC superfamily are primary transporters, the rest being secondary transporters utilizing proton or sodium gradient as a source of energy. Whereas MFS dominates in Gram positive bacteria , the RND family is unique to Gram-negatives.
Function
Although antibiotics are the most clinically important substrates of efflux systems, it is probable that most efflux pumps have other natural physiological functions. Examples include:
* The E. coli AcrAB efflux system, which has a physiologic role of pumping out bile acids and fatty acids to lower their toxicity.
* The MFS family Ptr pump in Streptomyces pristinaespiralis appears to be an autoimmunity pump for this organism when it turns on production of pristinamycins I and II.
* The AcrAB–TolC system in E.coli is suspected to have a role in the transport of the calcium-channel components in the E. coli membrane.
* The MtrCDE system plays a protective role by providing resistance to faecal lipids in rectal isolates of Neisseria gonorrhoeae.
* The AcrAB efflux system of Erwinia amylovora is important for this organism's virulence, plant (host) colonization, and resistance to plant toxins.
* The MexXY component of the MexXY-OprM multidrug efflux system of P. aeruginosa is inducible by antibiotics that target ribosomes via the PA5471 gene product[1].
The ability of efflux systems to recognize a large number of compounds other than their natural substrates is probably because substrate recognition is based on physicochemical properties, such as hydrophobicity, aromaticity and ionizable character rather than on defined chemical properties, as in classical enzyme-substrate or ligand-receptor recognition. Because most antibiotics are amphiphilic molecules - possessing both hydrophilic and hydrophobic characters - they are easily recognized by many efflux pumps.
Blastocyst Formation Video

The morula is a solid ball of about 16 undifferentiated, spherical cells. As cell division continues in the morula, the blastomeres change their shape and tightly align themselves against each other. This is called compaction and is likely mediated by cell surface adhesion glycoproteins.

CTLA-4 and Mechanism of action of anti-CTLA4

CTLA4 (Cytotoxic T-Lymphocyte Antigen 4) also known as CD152 (Cluster of differentiation 152) is a protein that plays an important regulatory role in the immune system. In humans, the CTLA4 protein is encoded by the CTLA4 gene.

Leading vs. Lagging strand replication
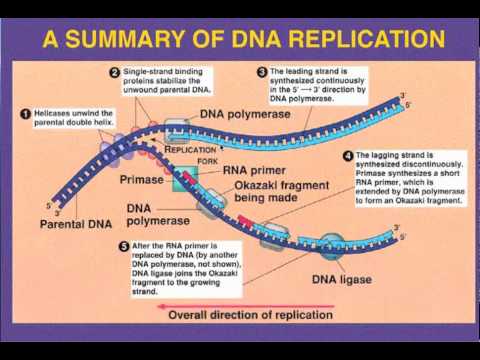
Leading strand
The leading strand is the template strand of the DNA double helix so that the replication fork moves along it in the 3' to 5' direction. This allows the newly synthesized strand complementary to the original strand to be synthesized 5' to 3' in the same direction as the movement of the replication fork.
On the leading strand, a polymerase "reads" the DNA and adds nucleotides to it continuously. This polymerase is DNA polymerase III (DNA Pol III) in prokaryotes and presumably Pol ε in yeasts. In human cells the leading and lagging strands are synthesized by Pol α and Pol δ within the nucleus and Pol γ in the mitochondria. Pol ε can substitute for Pol δ in special circumstances.

Lagging strand
The lagging strand is the strand of the template DNA double helix that is oriented so that the replication fork moves along it in a 5' to 3' manner. Because of its orientation, opposite to the working orientation of DNA polymerase III, which moves on a template in a 3' to 5' manner, replication of the lagging strand is more complicated than that of the leading strand.
On the lagging strand, primase "reads" the DNA and adds RNA to it in short, separated segments. In eukaryotes, primase is intrinsic to Pol α. DNA polymerase III or Pol δ lengthens the primed segments, forming Okazaki fragments. Primer removal in eukaryotes is also performed by Pol δ.[18] In prokaryotes, DNA polymerase I "reads" the fragments, removes the RNA using its flap endonuclease domain (RNA primers are removed by 5'-3' exonuclease activity of polymerase I [weaver, 2005], and replaces the RNA nucleotides with DNA nucleotides (this is necessary because RNA and DNA use slightly different kinds of nucleotides). DNA ligase joins the fragments together.
Avery–MacLeod–McCarty experiment

The Avery–MacLeod–McCarty experiment was an experimental demonstration, reported in 1944 by Oswald Avery, Colin MacLeod, and Maclyn McCarty, that DNA is the substance that causes bacterial transformation. It was the culmination of research in the 1930s and early 1940s at the Rockefeller Institute for Medical Research to purify and characterize the "transforming principle" responsible for the transformation phenomenon first described in Griffith's experiment of 1928: killed Streptococcus pneumoniae of the virulent strain type III-S, when injected along with living but non-virulent type II-R pneumococci, resulted in a deadly infection of type III-S pneumococci. In their paper "Studies on the Chemical Nature of the Substance Inducing Transformation of Pneumococcal Types: Induction of Transformation by a Deoxyribonucleic Acid Fraction Isolated from Pneumococcus Type III", published in the February 1944 issue of the Journal of Experimental Medicine, Avery and his colleagues suggest that DNA, rather than protein as widely believed at the time, may be the hereditary material of bacteria, and could be analogous to genes and/or viruses in higher organisms.
Nanomedicine Animation

Nanomedicine is the medical application of nanotechnology. It covers areas such as nanoparticle drug delivery and possible future applications of molecular nanotechnology (MNT) and nanovaccinology. Current problems for nanomedicine involve understanding the issues related to toxicity and environmental impact of nanoscale materials

Nanomedicine research is directly funded, with the US National Institutes of Health in 2005 funding a five-year plan to set up four nanomedicine centers. In April 2006, the journal Nature Materials estimated that 130 nanotech-based drugs and delivery systems were being developed worldwide
In the near future, advancement in nanomedicine will deliver a valuable set of research tools and clinically helpful devices. The National Nanotechnology Initiative expects new commercial applications in the pharmaceutical industry that will include advanced drug delivery systems, new therapies, and in vivo imaging. The most important innovations are taking place in drug delivery which involves developing nanoscale particles or molecules to improve bioavailability. Bioavailability refers to the presence of drug molecules where they are needed in the body and where they will do the most good. Drug delivery focuses on maximizing bioavailability both at specific places in the body and over a period of time. Over 65 billion dollars is wasted every year because of poor bioavailability. In vivo imaging is another area where tools and devices are being developed. Using nanoparticle contrast agents, images such as ultrasound and MRI have a favorable distribution and improved contrast. The new therapies and surgeries that are being developed might be effective in treating illnesses and diseases such as cancer. Finally, a shift from the possible to the potential will be made when nanorobots such as neuro-electronic interfaces and cell repair machines are discussed.
Drug delivery systems, lipid- or polymer-based nanoparticles, can be designed to improve the pharmacological and therapeutic properties of drugs. The strength of drug delivery systems is their ability to alter the pharmacokinetics and biodistribution of the drug. Nanoparticles have unusual properties that can be used to improve drug delivery. Where larger particles would have been cleared from the body, cells take up these nanoparticles because of their size. Complex drug delivery mechanisms are being developed, including the ability to get drugs through cell walls and into cells. Efficiency is important because many diseases depend upon processes within the cell and can only be impeded by drugs that make their way into the cell. Triggered response is one way for drug molecules to be used more efficiently. Drugs are placed in the body and only activate on encountering a particular signal. For example, a drug with poor solubility will be replaced by a drug delivery system where both hydrophilic and hydrophobic environments exist, improving the solubility. Also, a drug may cause tissue damage, but with drug delivery, regulated drug release can eliminate the problem. If a drug is cleared too quickly from the body, this could force a patient to use high doses, but with drug delivery systems clearance can be reduced by altering the pharmacokinetics of the drug. Poor biodistribution is a problem that can affect normal tissues through widespread distribution, but the particulates from drug delivery systems lower the volume of distribution and reduce the effect on non-target tissue. Potential nanodrugs will work by very specific and well-understood mechanisms, one of the major impacts of nanotechnology and nanoscience will be in leading development of completely new drugs with more useful behavior and less side effects.
Nanorobots
The somewhat speculative claims about the possibility of using nanorobots in medicine, advocates say, would totally change the world of medicine once it is realized. Nanomedicine would make use of these nanorobots, introduced into the body, to repair or detect damages and infections. A typical blood borne medical nanorobot would be between 0.5-3 micrometres in size, because that is the maximum size possible due to capillary passage requirement. Carbon would be the primary element used to build these nanorobots due to the inherent strength and other characteristics of some forms of carbon (diamond/fullerene composites). Cancer can be treated very effectively, according to nanomedicine advocates. Nanorobots could counter the problem of identifying and isolating cancer cells as they could be introduced into the blood stream. These nanorobots would search out cancer affected cells using certain molecular markers. Medical nanorobots would then destroy these cells, and only these cells. Nanomedicines could be a very helpful and hopeful therapy for patients, since current treatments like radiation therapy and chemotherapy often end up destroying more healthy cells than cancerous ones. From this point of view, it provides a non-depressed therapy for cancer patients. Nanorobots could also be useful in treating vascular disease, physical trauma , and even biological
Neuro-electronic Interfaces
Neuro-electronic interfaces are a visionary goal dealing with the construction of nanodevices that will permit computers to be joined and linked to the nervous system. This idea requires the building of a molecular structure that will permit control and detection of nerve impulses by an external computer. The computers will be able to interpret, register, and respond to signals the body gives off when it feels sensations. The demand for such structures is huge because many diseases involve the decay of the nervous system (ALS and multiple sclerosis). Also, many injuries and accidents may impair the nervous system resulting in dysfunctional systems and paraplegia. If computers could control the nervous system through neuro-electronic interface, problems that impair the system could be controlled so that effects of diseases and injuries could be overcome. Two considerations must be made when selecting the power source for such applications. They are refuelable and nonrefuelable strategies. A refuelable strategy implies energy is refilled continuously or periodically with external sonic, chemical, tethered, or electrical sources. A nonrefuelable strategy implies that all power is drawn from internal energy storage which would stop when all energy is drained.
One limitation to this innovation is the fact that electrical interference is a possibility. Electric fields, EMP pulses, and stray fields from other in vivo electrical devices can all cause interference. Also, thick insulators are required to prevent electron leakage, and if high conductivity of the in vivo medium occurs there is a risk of sudden power loss and “shorting out.” Finally, thick wires are also needed to conduct substantial power levels without overheating. Little practical progress has been made even though research is happening. The wiring of the structure is extremely difficult because they must be positioned precisely in the nervous system so that it is able to monitor and respond to nervous signals. The structures that will provide the interface must also be compatible with the body’s immune system so that they will remain unaffected in the body for a long time. In addition, the structures must also sense ionic currents and be able to cause currents to flow backward. While the potential for these structures is amazing, there is no timetable for when they will be available.
Cell repair machines
Using drugs and surgery, doctors can only encourage tissues to repair themselves. With molecular machines, there will be more direct repairs. Cell repair will utilize the same tasks that living systems already prove possible. Access to cells is possible because biologists can stick needles into cells without killing them. Thus, molecular machines are capable of entering the cell. Also, all specific biochemical interactions show that molecular systems can recognize other molecules by touch, build or rebuild every molecule in a cell, and can disassemble damaged molecules. Finally, cells that replicate prove that molecular systems can assemble every system found in a cell. Therefore, since nature has demonstrated the basic operations needed to perform molecular-level cell repair, in the future, nanomachine based systems will be built that are able to enter cells, sense differences from healthy ones and make modifications to the structure.
The possibilities of these cell repair machines are impressive. Comparable to the size of viruses or bacteria, their compact parts will allow them to be more complex. The early machines will be specialized. As they open and close cell membranes or travel through tissue and enter cells and viruses, machines will only be able to correct a single molecular disorder like DNA damage or enzyme deficiency. Later, cell repair machines will be programmed with more abilities with the help of advanced AI systems.
Nanocomputers will be needed to guide these machines. These computers will direct machines to examine, take apart, and rebuild damaged molecular structures. Repair machines will be able to repair whole cells by working structure by structure. Then by working cell by cell and tissue by tissue, whole organs can be repaired. Finally, by working organ by organ, health is restored to the body. Cells damaged to the point of inactivity can be repaired because of the ability of molecular machines to build cells from scratch. Therefore, cell repair machines will free medicine from reliance on self repair.
A new wave of technology and medicine is being created and its impact on the world is going to be monumental. From the possible applications such as drug delivery and in vivo imaging to the potential machines of the future, advancements in nanomedicine are being made every day. It will not be long for the 10 billion dollar industry to explode into a 100 billion or 1 trillion dollar industry, and drug delivery, in vivo imaging and therapy is just the beginning.
Source:Wikepedia
Function of the Neuromuscular Junction
A neuromuscular junction (NMJ) is the synapse or junction of the axon terminal of a motoneuron with the motor end plate, the highly-excitable region of muscle fiber plasma membrane responsible for initiation of action potentials across the muscle's surface, ultimately causing the muscle to contract. In vertebrates, the signal passes through the neuromuscular junction via the neurotransmitter acetylcholine.
Anatomy
When a motor neuron enters a muscle, it loses its myelin sheath and splits into many terminal branches. Motor neuron (efferent) axons originating in the spinal cord enter muscle fibers, where they split into many unmyelinated branches. These terminal fibers run along the myocytes to end at the neuromuscular junction, which occupies a depression in the sarcolemma. Each motor neuron can innervate from one to over 2000 muscle fibers, but each muscle fiber receives inputs from only one motor neuron.
In the terminal bouton of the motor nerve, structures known as presynaptic active zones accumulate synaptic vesicles filled with the neurotransmitter acetylcholine.
On the muscle side of the junction, the muscle fiber is folded into grooves called postjunctional folds that mirror the presynaptic active zones, the spaces between the folds contain acetylcholine receptors.
The muscle surface is covered by the synaptic basal lamina. Postjunctional folds are characteristic of skeletal muscle, particularly in fast muscle fibers.
Mechanism of actionUpon the arrival of an action potential at the axon terminal, voltage-dependent calcium channels open and Ca2+ ions flow from the extracellular fluid into the motor neuron's cytosol. This influx of Ca2+ triggers excitation-contraction coupling, a biochemical cascade that causes neurotransmitter-containing vesicles to fuse to the motor neuron's cell membrane and release acetylcholine into the synaptic cleft.
Acetylcholine diffuses across the synaptic cleft and binds to the nicotinic acetylcholine receptors that dot the motor end plate.
The receptors are ligand-gated ion channels, and when bound by acetylcholine, they open, allowing sodium and potassium ions to flow in and out of the muscle's cytosol, respectively.
Because of the differences in electrochemical gradients across the plasma membrane, more sodium moves in than potassium out, producing a local depolarization of the motor end plate known as an end-plate potential (EPP).
This depolarization spreads across the surface of the muscle fiber into transverse tubules, eliciting the release of calcium from the sarcoplasmic reticulum, thus initiating muscle contraction.
The action of acetylcholine is terminated when the enzyme acetylcholinesterase degrades the neurotransmitter and the unhydrolysed neurotransmitter diffuses away.

HIV Replication Animation

Entry to the cell
 Subscribe in a reader
Subscribe in a reader
HIV enters macrophages and CD4+ T cells by the adsorption of glycoproteins on its surface to receptors on the target cell followed by fusion of the viral envelope with the cell membrane and the release of the HIV capsid into the cell.
 Subscribe in a reader
Subscribe in a reader Entry to the cell begins through interaction of the trimeric envelope complex (gp160 spike, discussed above) and both CD4 and a chemokine receptor (generally either CCR5 or CXCR4, but others are known to interact) on the cell surface. The gp160 spike contains binding domains for both CD4 and chemokine receptors. The first step in fusion involves the high-affinity attachment of the CD4 binding domains of gp120 to CD4. Once gp120 is bound with the CD4 protein, the envelope complex undergoes a structural change, exposing the chemokine binding domains of gp120 and allowing them to interact with the target chemokine receptor. This allows for a more stable two-pronged attachment, which allows the N-terminal fusion peptide gp41 to penetrate the cell membrane.Repeat sequences in gp41, HR1 and HR2 then interact, causing the collapse of the extracellular portion of gp41 into a hairpin. This loop structure brings the virus and cell membranes close together, allowing fusion of the membranes and subsequent entry of the viral capsid.
Once HIV has bound to the target cell, the HIV RNA and various enzymes, including reverse transcriptase, integrase, ribonuclease and protease, are injected into the cell. During the microtubule based transport to the nucleus, the viral single strand RNA genome is transcribed into double strand DNA, which is then integrated into a host chromosome.
HIV can infect dendritic cells (DCs) by this CD4-CCR5 route, but another route using mannose-specific C-type lectin receptors such as DC-SIGN can also be used. DCs are one of the first cells encountered by the virus during sexual transmission. They are currently thought to play an important role by transmitting HIV to T cells once the virus has been captured in the mucosa by DCs.
Replication and transcription Once the viral capsid enters the cell, an enzyme called reverse transcriptase liberates the single-stranded (+)RNA from the attached viral proteins and copies it into a complementary DNA.This process of reverse transcription is extremely error-prone and it is during this step that mutations may occur. Such mutations may cause drug resistance. The reverse transcriptase then makes a complementary DNA strand to form a double-stranded viral DNA intermediate (vDNA). This vDNA is then transported into the cell nucleus. The integration of the viral DNA into the host cell's genome is carried out by another viral enzyme called integrase.
This integrated viral DNA may then lie dormant, in the latent stage of HIV infection. To actively produce the virus, certain cellular transcription factors need to be present, the most important of which is NF-κB (NF kappa B), which is upregulated when T cells become activated. This means that those cells most likely to be killed by HIV are those currently fighting infection.
Rev-mediated HIV mRNA transport. Rev (red) binds the Rev response element (RRE, blue) to mediate export of unspliced and singly spliced mRNA from the nucleus to the cytoplasm. In this replication process, the integrated provirus is copied to mRNA which is then spliced into smaller pieces. These small pieces produce the regulatory proteins Tat (which encourages new virus production) and Rev. As Rev accumulates it gradually starts to inhibit mRNA splicing. At this stage, the structural proteins Gag and Env are produced from the full-length mRNA. The full-length RNA is actually the virus genome; it binds to the Gag protein and is packaged into new virus particles.
HIV-1 and HIV-2 appear to package their RNA differently; HIV-1 will bind to any appropriate RNA whereas HIV-2 will preferentially bind to the mRNA which was used to create the Gag protein itself. This may mean that HIV-1 is better able to mutate (HIV-1 infection progresses to AIDS faster than HIV-2 infection and is responsible for the majority of global infections).
Assembly and release The final step of the viral cycle, assembly of new HIV-1 virons, begins at the plasma membrane of the host cell. The Env polyprotein (gp160) goes through the endoplasmic reticulum and is transported to the Golgi complex where it is cleaved by protease and processed into the two HIV envelope glycoproteins gp41 and gp120. These are transported to the plasma membrane of the host cell where gp41 anchors the gp120 to the membrane of the infected cell. The Gag (p55) and Gag-Pol (p160) polyproteins also associate with the inner surface of the plasma membrane along with the HIV genomic RNA as the forming virion begins to bud from the host cell. Maturation either occurs in the forming bud or in the immature virion after it buds from the host cell. During maturation, HIV proteases cleave the polyproteins into individual functional HIV proteins and enzymes. The various structural components then assemble to produce a mature HIV virion. This cleavage step can be inhibited by protease inhibitors. The mature virus is then able to infect another cell.
Gleevecs Effects on tryosine Kinase
.jpg)
Imatinib is a drug used to treat certain types of cancer. It is currently marketed by Novartis as Gleevec (USA) or Glivec (Europe/Australia) as its mesylate salt, imatinib mesilate (INN). It was originally coded during development as CGP57148B or STI-571 (these terms are used in early preclinical publications). It is used in treating chronic myelogenous leukemia (CML), gastrointestinal stromal tumors (GISTs) and a number of other malignancies.
The animation begins by introducing the Philadelphia Chromosome, the result of a reciprocal translocation between chromosomes 9 and 22. More specifically the breakpoint cluster region (BCR) of chromosome 22 is fused with part of the Abelson (ABL) gene on chromosome 9. The resulting BCR-ABL genetic domain now located within chromosome 22 and codes for a mutant tyrosine kinase also known as BCR-ABL. Under normal circumstances tyrosine kinase proteins respond to external cellular messaging proteins, and ultimately initiate a series of reactions that culminate in cellular replication.
Conversely, BCR-ABL is constitutively active, meaning it does not require activation by the aforementioned cellular messaging proteins in order to stimulate cellular replication. This results in acceleration of cell division, an inhibition of DNA repair, overall genomic instability, and the fatal blast crisis characteristic of chronic myelogenous leukemia. The animation progresses to introduce Gleevec (imatinib), the first in a class of drugs that specifically target and competively inhibit the ATP binding site on BCR-ABL tyrosine kinase. This prevents the ABL domain from phosphorylating the tyrosine residue, and as a result preventing the proliferation of hematopoietic cells that express BCR-ABL. Therapy with imatinib results in a dramatic reduction of tumor clone cells and the occurrence of blast crisis', through targeted drug treatment which leaves health cells unscathed.
.jpg)







Protein Synthesis Animation

Protein biosynthesis (Synthesis) is the process in which cells build proteins. The term is sometimes used to refer only to protein translation but more often it refers to a multi-step process, beginning with amino acid synthesis and transcription which are then used for translation. Protein biosynthesis, although very similar, differs between prokaryotes and eukaryotes.
Amino acid synthesis
Amino acids are the monomers which are polymerized to produce proteins. Amino acid synthesis is the set of biochemical processes (metabolic pathways) which build the amino acids from carbon sources like glucose. Not all amino acids may be synthesised by every organism, for example adult humans have to obtain 8 of the 20 amino acids from their diet.
The amino acids are then loaded onto tRNA molecules for use in the process of translation.
Transcription is the process by which an mRNA template, carrying the sequence of the protein, is produced for the translation step from the genome. Transcription makes the template from one strand of the DNA double helix, called the template strand. Transcription takes place in 3 stages.
Transcription starts with the process of initiation. RNA polymerase, the enzyme which produces RNA from a DNA template, binds to a specific region on DNA that designates the starting point of transcription. This binding region is called the promoter. As the RNA polymerase binds on to the promoter, the DNA strands are beginning to unwind.
The second process is elongation. RNA polymerase travels along the template (noncoding) strand, synthesizing a ribonucleotide polymer. RNA polymerase does not use the coding strand as a template because a copy of any strand produces a base sequence that is complementary to the strand which is being copied. Therefore DNA from the noncoding strand is used as a template to copy the coding strand.
The third stage is termination. As the polymerase reaches the termination stage, modifications are required for the newly transcribed mRNA to be able to travel to the other parts of the cell, including cytoplasm and endoplasmic reticulum, for translation. A 5' cap is added to the mRNA to protect it from degradation. In eukaryotes, poly-A-polymerase adds a poly-A tail onto the 3’ end for stabilization, protection from cytoplasmic hydrolytic enzymes, and as a template for further processes. Also in eukaryotes (higher organisms) the vital process of splicing occurs at this stage by the spliceosome enzyme. It removes the introns (non-coding bits of genetic material) and glues together the exons (the segments that code for a specific protein).
The mRNA now exits the nuclear pore to be translated.
Translation
Protein translation involves the transfer of information from the mRNA into a peptide, composed of amino acids. This process is mediated by the ribosome, with the adaptation of the RNA sequence into amino acids mediated by transfer RNA. Numerous initation and elongation factors also play a role.
Translation requires a lot of energy, with the hydrolysis of approximately 4 NTP --> NDP per amino acid added. (This includes the aminoacylation of the tRNA. Thus, gene expression is highly regulated to ensure that only proteins that are required are translated.
Translation involves 3 processes: initiation, elongation, and termination.
Initiation in Prokaryotes
The initiation of protein translation involves the assembly of the ribosome and addition of the first amino acid, methionine.
The 30S ribosomal subunit attaches to the mRNA, mediated by IF-1 and IF-3 (initiation factors). The 30S ribosome brings with it the P and A site, but the A site is blocked by IF-1 to prevent binding of tRNA. It aligns to the Shine-Dalgarno sequence, which positions the first codon (AUG) in the P site.
Next, the specific aminoacyl-tRNA for N-formylmethionine (F-Met) is brought into the P site by IF-2. The anticodon of this tRNA will bind to the AUG codon on the mRNA. Note: this is the only tRNA brought into the P site; all successive aminoacyl-tRNAs will be brought to the A site for peptide elongation.
The 50S ribosomal subunit is then brought in to complete the ribosome, and with it, IF-1, IF-2, and IF-3 come off the complex. The A and P site are completed, and the 50S subunit also brings the E (exit) site.
Initiation in Eukaryotes
The initiation of protein translation in eukaryotes is similar to that of prokaryotes with some modifications.
A complex of proteins will connect the 5'cap and 3'PolyA tail, and this complex will recruit the ribosome subunits.
There is no Shine-Dalgarno sequence in eukaryotes. Instead, the ribosome scans along the mRNA for the first methionine codon. Similarly, there is no N-formylmethionine in eukaryotic cells.
Elongation
Elongation of protein biosynthesis is fairly similar between prokaryotes and eukaryotes. The following is a description of elongation in prokaryotes.
Elongation proceeds after initiation with the binding of an aminoacyl-tRNA to the A site, which is the next codon in the mRNA. The aminoacyl-tRNA is brought to the ribosome through a series of interactions with EF-Tu (an elongation factor). This step involves the hydrolysis of GTP: EF-Tu-GTP --> EF-Tu-GDP (The hydrolyzed GDP is switched for GTP through another series of reactions with EF-Ts.)
The next aminoacyl-tRNA binds to the codon, and the C-terminus of the F-Met undergoes nucleophilic attack by the N-terminus of the second amino acid. The F-Met is now connected to the second amino acid through a peptide bond.
The first tRNA (for F-Met) is now uncharged. The entire ribosome complex moves along the mRNA through the action of another elongation factor (EF-G) and the hydrolysis of GTP --> GDP.
The first tRNA is now in the E site and comes off from the ribosome, while the second tRNA, with the nascent peptide chain, is in the P site. Step 1-4 will repeat as successive amino acids are added.
Termination
Termination of protein biosynthesis occurs when the ribosome comes across a stop codon, for which there is no tRNA. At this point, protein biosynthesis halts and one of three release factors will bind to the stop codon. (Note: In eukaryotes, there is only one release factor that will bind to all three stop codons.) This induces a nucleophilic attack of the C-terminus of the nascent peptide by water - this hydrolysis releases the peptide from the ribosome. The ribosome, release factor, and uncharged tRNA then dissociates and translation is complete.
Events following Protein Translation
The events following biosynthesis include post-translational modification and protein folding. During and after synthesis, polypeptide chains often fold to assume, so called, native secondary and tertiary structures. This is known as protein folding.
Many proteins undergo post-translational modification. This may include the formation of disulfide bridges or attachment of any of a number of biochemical functional groups, such as acetate, phosphate, various lipids and carbohydrates. Enzymes may also remove one or more amino acids from the leading (amino) end of the polypeptide chain, leaving a protein consisting of two polypeptide chains connected by disulfide bonds.
Actin 4 Binding region

Alpha actinins belong to the spectrin gene superfamily which represents a diverse group of cytoskeletal proteins, including the alpha and beta spectrins and dystrophins. Alpha actinin is an actin-binding protein with multiple roles in different cell types. In nonmuscle cells, the cytoskeletal isoform is found along microfilament bundles and adherens-type junctions, where it is involved in binding actin to the membrane. In contrast, skeletal, cardiac, and smooth muscle isoforms are localized to the Z-disc and analogous dense bodies, where they help anchor the myofibrillar actin filaments. This gene encodes a nonmuscle, alpha actinin isoform which is concentrated in the cytoplasm, and thought to be involved in metastatic processes. Mutations in this gene have been associated with focal and segmental glomerulosclerosis.
Asparaginase

Asparaginase is an enzyme that catalyzes the hydrolysis of asparagine to aspartic acid. It is marketed under the brand name Elspar, to treat acute lymphoblastic leukemia (ALL) and is also used in some mast cell tumor protocols. Unlike other chemotherapy agents, it can be given as an intramuscular, subcutaneous, or intravenous injection without fear of tissue irritation.
 Mechanism of action
Mechanism of action
The rationale behind asparaginase is that it takes advantage of the fact that ALL leukemic cells are unable to synthesize the non-essential amino acid asparagine, whereas normal cells are able to make their own asparagine; thus leukemic cells require high amount of asparagine. These leukemic cells depend on circulating asparagine. Asparaginase, however, catalyzes the conversion of L-asparagine to aspartic acid and ammonia. This deprives the leukemic cell of circulating asparagine.
Vaccines Animation

Vaccines is a biological preparation that improves immunity to a particular disease. A vaccine typically contains a small amount of an agent that resembles a microorganism. The agent stimulates the body's immune system to recognize the agent as foreign, destroy it, and "remember" it, so that the immune system can more easily recognize and destroy any of these microorganisms that it later encounters.
Vaccines can be prophylactic (e.g. to prevent or ameliorate the effects of a future infection by any natural or "wild" pathogen), or therapeutic (e.g. vaccines against cancer are also being investigated; see cancer vaccine).

Types
Vaccines are dead or inactivated organisms or purified products derived from them.
There are several types of vaccines currently in use.These represent different strategies used to try to reduce risk of illness, while retaining the ability to induce a beneficial immune response.
Killed
Vaccines containing killed microorganisms - these are previously virulent micro-organisms which have been killed with chemicals or heat. Examples are vaccines against flu, cholera, bubonic plague, polio and hepatitis A.
Attenuated
Some vaccines contain live, attenuated virus microorganisms. These are live micro-organisms that have been cultivated under conditions that disable their virulent properties, or which use closely-related but less dangerous organisms to produce a broad immune response. They typically provoke more durable immunological responses and are the preferred type for healthy adults. Examples include yellow fever, measles, rubella, and mumps. The live Mycobacterium tuberculosis vaccine developed by Calmette and Guérin is not made of a contagious strain, but contains a virulently modified strain called "BCG" used to elicit immunogenicity to the vaccine.
Toxoid
Toxoids - these are inactivated toxic compounds in cases where these (rather than the micro-organism itself) cause illness. Examples of toxoid-based vaccines include tetanus and diphtheria. Not all toxoids are for micro-organisms; for example, Crotalus atrox toxoid is used to vaccinate dogs against rattlesnake bites.
Subunit
Protein subunit - rather than introducing an inactivated or attenuated micro-organism to an immune system (which would constitute a "whole-agent" vaccine), a fragment of it can create an immune response. Characteristic examples include the subunit vaccine against Hepatitis B virus that is composed of only the surface proteins of the virus (produced in yeast) and the virus-like particle (VLP) vaccine against human papillomavirus (HPV) that is composed of the viral major capsid protein.
Conjugate
Conjugate - certain bacteria have polysaccharide outer coats that are poorly immunogenic. By linking these outer coats to proteins (e.g. toxins), the immune system can be led to recognize the polysaccharide as if it were a protein antigen. This approach is used in the Haemophilus influenzae type B vaccine.
Experimental
A number of innovative vaccines are also in development and in use:
* Recombinant Vector - by combining the physiology of one micro-organism and the DNA of the other, immunity can be created against diseases that have complex infection processes
* DNA vaccination - in recent years a new type of vaccine called DNA vaccination, created from an infectious agent's DNA, has been developed. It works by insertion (and expression, triggering immune system recognition) of viral or bacterial DNA into human or animal cells. Some cells of the immune system that recognize the proteins expressed will mount an attack against these proteins and cells expressing them. Because these cells live for a very long time, if the pathogen that normally expresses these proteins is encountered at a later time, they will be attacked instantly by the immune system. One advantage of DNA vaccines is that they are very easy to produce and store. As of 2006, DNA vaccination is still experimental.
* T-cell receptor peptide vaccines are under development for several diseases using models of Valley Fever, stomatitis, and atopic dermatitis. These peptides have been shown to modulate cytokine production and improve cell mediated immunity.
* Targeting of identified bacterial proteins that are involved in complement inhibition would neutralize the key bacterial virulence mechanism.
While most vaccines are created using inactivated or attenuated compounds from micro-organisms, synthetic vaccines are composed mainly or wholly of synthetic peptides, carbohydrates or antigens.
Valence
Vaccines may be monovalent (also called univalent) or multivalent (also called polyvalent). A monovalent vaccine is designed to immunize against a single antigen or single microorganism. A multivalent or polyvalent vaccine is designed to immunize against two or more strains of the same microorganism, or against two or more microorganisms. In certain cases a monovalent vaccine may be preferable for rapidly developing a strong immune response.
Vaccine. (2009, November 8). In Wikipedia, The Free Encyclopedia. Retrieved 07:59, November 9, 2009, from http://en.wikipedia.org/w/index.php?title=Vaccine&oldid=324652602
Bacteriorhodopsin

Bacteriorhodopsin is a protein used by archaea, most notably halobacteria. It acts as a proton pump, i.e. it captures light energy and uses it to move protons across the membrane out of the cell. The resulting proton gradient is subsequently converted into chemical energy.

Bacteriorhodopsin is an integral membrane protein usually found in two-dimensional crystalline patches known as "purple membrane", which can occupy up to nearly 50% of the surface area of the archaeal cell. The repeating element of the hexagonal lattice is composed of three identical protein chains, each rotated by 120 degrees relative to the others. Each chain has seven transmembrane alpha helices and contains one molecule of retinal buried deep within, the typical structure for retinylidene proteins.
It is the retinal molecule that changes its conformation when absorbing a photon, resulting in a conformational change of the surrounding protein and the proton pumping action. It is covalently linked to Lys216 in the chromophore by Schiff base action. After photoisomerization of the retinal molecule, Asp85 becomes a proton acceptor of the donor proton from the retinal molecule. This releases a proton from a "holding site" into the extracellular side (EC) of the membrane. Reprotonation of the retinal molecule by Asp96 restores its original isomerized form. This results in a second proton being released to the EC side. Asp85 releases its proton into the "holding site" where a new cycle may begin.
The bacteriorhodopsin molecule is purple and is most efficient at absorbing green light (wavelength 500-650 nm, with the absorption maximum at 568 nm).
The three-dimensional tertiary structure of bacteriorhodopsin resembles that of vertebrate rhodopsins, the pigments that sense light in the retina. Rhodopsins also contain retinal; however, the functions of rhodopsin and bacteriorhodopsin are different and there is only slight homology in their amino acid sequences. Both rhodopsin and bacteriorhodopsin belong to the 7TM receptor family of proteins, but rhodopsin is a G protein coupled receptor and bacteriorhodopsin is not. In the first use of electron crystallography to obtain an atomic-level protein structure, the structure of bacteriorhodopsin was resolved in 1990. It was then used as a template to build models of other G protein-coupled receptors before crystallographic structures were also available for these proteins.
Many molecules have homology to bacteriorhodopsin, including the light-driven chloride pump halorhodopsin (for whom the crystal structure is also known), and some directly light-activated channels like channelrhodopsin.
All other photosynthetic systems in bacteria, algae and plants use chlorophylls or bacteriochlorophylls rather than bacteriorhodopsin. These also produce a proton gradient, but in a quite different and more indirect way involving an electron transfer chain consisting of several other proteins. Furthermore, chlorophylls are aided in capturing light energy by other pigments known as "antennas"; these are not present in bacteriorhodopsin based systems. Lastly, chlorophyll-based photosynthesis is coupled to carbon fixation (the incorporation of carbon dioxide into larger organic molecules); this is not true for bacteriorhodopsin-based system. It is thus likely that photosynthesis independently evolved at least twice, once in bacteria and once in archaea.
"Bacteriorhodopsin." Wikipedia, The Free Encyclopedia. 23 Aug 2009, 19:24 UTC. 21 Oct 2009 <http://en.wikipedia.org/w/index.php?title=Bacteriorhodopsin&oldid=309646691>.
"Bacteriorhodopsin." Wikipedia, The Free Encyclopedia. 23 Aug 2009, 19:24 UTC. 21 Oct 2009 <http://en.wikipedia.org/w/index.php?title=Bacteriorhodopsin&oldid=309646691>.

Preimplantation Genetic Diagnosis

Preimplantation genetic diagnosis (PGD or PIGD) (also known as embryo screening) refers to procedures that are performed on embryos prior to implantation, sometimes even on oocytes prior to fertilization. PGD is considered another way to prenatal diagnosis. Its main advantage is that it avoids selective pregnancy termination as the method makes it highly likely that the baby will be free of the disease under consideration. PGD thus is an adjunct to assisted reproductive technology, and requires in vitro fertilization (IVF) to obtain oocytes or embryos for evaluation.
Indications and applications
Currently, there are mainly two groups of patients for which PGD is being applied:
In the first group PGD is used to look for a specific disorder in couples with a high risk of transmitting an inherited condition. This can be a monogenic disorder, meaning the condition is due to a single gene only, (autosomal recessive, autosomal dominant or X-linked disorders) or a chromosomal structural aberration (such as a balanced translocation). PGD helps these couples identify embryos carrying a genetic disease or a chromosome abnormality, thus avoiding the difficult choice of abortion. In addition, there are infertile couples who carry an inherited condition and who opt for PGD as it can be easily combined with their IVF treatment.
The second group consists of couples who undergo IVF treatment and whose embryos are screened for chromosome aneuploidies. The technique is not used to obtaining a specific prenatal diagnosis but rather for screening, properly referred to as preimplantation genetic screening (PGS), to increase the chances of an ongoing pregnancy. The main applications for PGS are an advanced maternal age, a history of recurrent miscarriages or repeated unsuccessful implantation. As the results of PGS rely on the assessment of a single cell, PGS has inherent limitations as the tested cell may not be representative of the embryo and embryo mosaicism may not be be clinically significant. Further, studies have not shown that IVF success rates in terms of live births are better when PGS is used, and there is some concern that a biopsy may lower success rates. It has also been proposed for patients with obstructive and non-obstructive azoospermia.

Nucleotide excision repair NER of carcinogen adducts

Nucleotide excision repair is a DNA repair mechanism. DNA constantly requires repair due to damage that can occur to bases from a vast variety of sources including chemicals, radiation and other mutagens. Nucleotide excision repair (NER) is a particularly important mechanism by which the cell can prevent unwanted mutations by removing the vast majority of UV-induced DNA damage (mostly in the form of thymine dimers and 6-4-photoproducts). The importance of this repair mechanism is evidenced by the severe human diseases that result from in-born genetic mutations of NER proteins including Xeroderma pigmentosum and Cockayne's syndrome. While the base excision repair machinery can recognize specific lesions in the DNA it can correct only damaged bases that can be removed by a specific glycosylase, the nucleotide excision repair enzymes recognize bulky distortions in the shape of the DNA double helix. Recognition of these distortions leads to the removal of a short single-stranded DNA segment that includes the lesion, creating a single-strand gap in the DNA, which is subsequently filled in by DNA polymerase, which uses the undamaged strand as a template. NER can be divided into two subpathways (Global genomic NER and Transcription coupled NER) that differ only in their recognition of helix-distorting DNA damage.

Enzyme Kinetics

Enzyme kinetics is the study of the chemical reactions that are catalysed by enzymes, with a focus on their reaction rates. The study of an enzyme's kinetics reveals the catalytic mechanism of this enzyme, its role in metabolism, how its activity is controlled, and how a drug or a poison might inhibit the enzyme.

Enzymes are usually protein molecules that manipulate other molecules — the enzymes' substrates. These target molecules bind to an enzyme's active site and are transformed into products through a series of steps known as the enzymatic mechanism. These mechanisms can be divided into single-substrate and multiple-substrate mechanisms. Kinetic studies on enzymes that only bind one substrate, such as triosephosphate isomerase, aim to measure the affinity with which the enzyme binds this substrate and the turnover rate.
Flagellum Motar

The flagellum is linked to a flexible hook. The hook is attached to a series of protein rings , which are embedded in the outer and inner (plasma) membranes. The rings form a rotor, which rotates with the flagellum at more than 100 revolutions per second. The rotation is driven by a flow of protons through an outer ring of proteins , the stator, which also contains the proteins responsible for switching the direction of rotation

LTP Mechanisms

Long-term potentiation (LTP) is the long-lasting improvement in communication between two neurons that results from stimulating them simultaneously. Since neurons communicate via chemical synapses, and because memories are believed to be stored within these synapses, LTP is widely considered one of the major cellular mechanisms that underlies learning and memory.
LTP shares many features with long-term memory that make it an attractive candidate for a cellular mechanism of learning. For example, LTP and long-term memory are triggered rapidly, each depends upon the synthesis of new proteins, each has properties of associativity, and each can potentially last for many months. LTP may account for many types of learning, from the relatively simple classical conditioning present in all animals, to the more complex, higher-level cognition observed in humans.
By enhancing synaptic transmission, LTP improves the ability of two neurons, one presynaptic and the other postsynaptic, to communicate with one another across a synapse. The precise mechanisms for this enhancement of transmission have not been fully established, in part because LTP is governed by multiple mechanisms that vary by such things as brain region, animal age, and species. Yet in the most well understood form of LTP, enhanced communication is predominantly carried out by improving the postsynaptic cell's sensitivity to signals received from the presynaptic cell. These signals, in the form of neurotransmitter molecules, are received by neurotransmitter receptors present on the surface of the postsynaptic cell. LTP improves the postsynaptic cell's sensitivity to neurotransmitter in large part by increasing the activity of existing receptors and by increasing the number of receptors on the postsynaptic cell surface.
LTP was discovered in the rabbit hippocampus by Terje Lømo in 1966 and has remained a popular subject of research since. Most modern LTP studies seek to better understand its basic biology, while others aim to draw a causal link between LTP and behavioral learning. Still others try to develop methods, pharmacologic or otherwise, of enhancing LTP to improve learning and memory. LTP is also a subject of clinical research, for example, in the areas of Alzheimer's disease and addiction medicine.
Mechanism
Long-term potentiation occurs through a variety of mechanisms throughout the nervous system; no single mechanism unites all of LTP's many types. However, for the purposes of study, LTP is commonly divided into three phases that occur sequentially: short-term potentiation, early LTP, and late LTP. Little is known about the mechanisms of short-term potentiation,
Each phase of LTP is governed by a set of mediators, small molecules that dictate the events of that phase. These molecules include protein receptors that respond to events outside of the cell, enzymes that carry out chemical reactions within the cell, and signaling molecules that allow the progression from one phase to the next. In addition to these mediators, there are also modulator molecules, described later, that interact with mediators to finely alter the LTP ultimately generated.
The early (E-LTP) and late (L-LTP) phases of LTP are each characterized by a series of three events: induction, maintenance, and expression. Induction is the process by which a short-lived signal triggers that phase of LTP to begin. Maintenance corresponds to the persistent biochemical changes that occur in response to the induction of that phase. Expression entails the long-lasting cellular changes that result from activation of the maintenance signal. Thus the mechanisms of LTP can be discussed in terms of the mediators that underlie the induction, maintenance, and expression of E-LTP and L-LTP.

Genome editing with engineered nucleases

Genome editing with engineered nucleases (GEEN) refers to a reverse genetics method using ‘molecular scissors’, or artificially engineered nucleases, to cut and create specific double-stranded break (DSB) at desired locations in the genome, harnessing the cell’s endogenous mechanisms to repair the induced break by natural processes of homologous recombination (HR) and nonhomologous end-joining (NHEJ). There are currently 3 families of engineered nucleases being used: Zinc Finger Nuclease (ZFN), Transcription Activator-Like Effector Nuclease (TALENs), and engineered meganuclease re-engineered homing endonucleases.
It is commonly practiced in genetic analysis that in order to understand the function of a gene or a protein function one interferes with it in a sequence-specific way and monitors its effects on the organism. However, in some organisms it is difficult or impossible to perform site-specific mutagenesis, and therefore more indirect methods have to be used, such as silencing the gene of interest by short RNA interference (siRNA) . Yet gene disruption by siRNA can be variable and incomplete. Genome editing with nucleases such as ZFN is different from siRNA in that the engineered nuclease is able to modify DNA-binding specificity and therefore can in principle cut any targeted position in the genome, and introduce modification of the endogenous sequences for genes that are impossible to specifically target by conventional RNAi. Furthermore, the specificity of ZFNs and TALENs are enhanced as two ZFNs are required in the recognition of their portion of the target and subsequently direct to the neighboring sequences.
It was chosen by Nature Methods as the 2011 Method of the Year.
Concept
Modern biology has greatly benefited from reverse genetics: the ability to start from particular genotypes and then look at the resultant phenotypes. Given that phenotypic changes are often complex and a result of multiple genetic interactions reverse genetics has been particularly significant in modern biology because of its inherent simpler nature. Among the key aspects of reverse genetic analysis is the ability to modify the genetic code. Although techniques such as site-directed mutagenesis allow such studies more in vitro settings, in vivo observations of phenotypic effects of the genetic changes provide a more comprehensive view of mutational significance. This has been made possible in yeast and mice by recombination based methods, which use the recombination machinery of cells to exchange DNA between a naturally occurring gene in the organism and an exogenous DNA source with the desired characteristics. However such techniques are less successful in other organisms and additionally require stringent selection steps and thus addition of selection specific sequences to the incorporated into the DNA which are a deviation from the naturally occurring genetic sequences. Furthermore they can be quite inefficient as only 1 of a million mouse embryonic stem cells treated with donor DNA incorporated it at the target sequence. Use of other techniques such as P-element transgenesis in Drosophila also have their limitations, the major one being the randomness of incorporation and the possibility of affecting other genes and expression patterns. However, genomic editing with engineered nucleases is a rapidly growing technology with the promise of overcoming these shortcomings by the use of relatively simple concepts.
Double stranded breaks and their repair
First and foremost in understanding the use of nucleases in genome editing is the understanding of DNA double stranded break (DSB) repair mechanisms. Two of the known DSB repair pathways that are essentially functional in all organisms are the non-homologous end joining (NHEJ) and homology directed repair (HDR). NHEJ uses a variety of enzymes to directly join the DNA ends in a DSB while in HDR, a homologous sequence is utilized as a template for regeneration of missing DNA sequence at the break point. The natural properties of these pathways form the very basis of nucleases based genome editing. NHEJ is error prone such that it was shown to cause mutations at the repair site in approximately 50% of DSB in mycobacteria and also its low fidelity has been linked to mutational accumulation in leukemias. Thus if one is able to create a DSB at a desired gene in multiple samples, it is very likely that mutations will be generated at that site in some of the treatments because of errors created by the NHEJ infidelity. On the other hand, the dependency of HDR on a homologous sequence to repair DSBs can be exploited by inserting a desired sequence within a sequence that is homologous to the flanking sequences of a DSB which, when used as a template by HDR system, would lead to the creation of the desired change within the genomic region of interest. Despite the distinct mechanisms, the concept of the HDR based gene editing is in a way similar to that of homologous recombination based gene targeting. However, the rate of recombination is increased by at least three orders of magnitude when DSBs are created and HDR is at work thus making the HDR based recombination much more efficient and eliminating the need for stringent positive and negative selection steps. So based on these principles if one is able to create a DSB at a specific location within the genome, then the cell’s own repair systems will help in creating the desired mutations.
Site-specific double stranded breaks
Creation of a DSB in DNA should not be a challenging task as the commonly used restriction enzymes are capable of doing so. However, if genomic DNA is treated with a particular restriction endonuclease many DSBs will be created. This is a result of the fact that most restriction enzymes recognize a few base pairs on the DNA as their target and very likely that particular base pair combination will be found in many locations across the genome. To overcome this challenge and create site-specific DSB, three distinct classes of nucleases have been discovered and bioengineered to date. These are the Zinc finger nucleases (ZFNs), transcription-activator like effector nucleases (TALENs) and meganucleases. Here we provide a brief overview and comparison of these enzymes and the concept behind their development.
Current engineered nucleases
Meganucleases, found commonly in microbial species, have the unique property of having very long recognition sequences (>14bp) thus making them naturally very specific. This can be exploited to make site-specific DSB in genome editing; however, the challenge is that not enough meganucleases are known, or may ever be known, to cover all possible target sequences. To overcome this challenge, mutagnesis and high throughput screening methods have been used to create megnuclease variants that recognize unique sequences. Others have been able to fuse various meganucleases and create hybrid enzymes that recognize a new sequence. Yet others have attempted to alter the DNA interacting aminoacids of the meganuclease to design sequence specific meganucelases in a method named rationally designed meganuclease (US Patent 8,021,867 B2).
Meganuclease have the benefit of causing less toxicity in cells compared to methods such as ZFNs likely because of more stringent DNA sequence recognition; however, the construction of sequence specific enzymes for all possible sequences is costly and time consuming as one is not benefitting from combinatorial possibilities that methods such as ZFNs and TALENs utilize. So there are both advantages and disadvantages.
As opposed to meganucleases, the concept behind ZFNs and TALENs is more based on a non-specific DNA cutting enzyme which would then be linked to specific DNA sequence recognizing peptides such as zinc fingers and transcription activator-like effectors (TALEs). The key to this was to find an endonuclease whose DNA recognition site and cleaving site were separate from each other, a situation that is not common among restriction enzymes. Once this enzyme was found, its cleaving portion could be separated which would be very non-specific as it would have no recognition ability. This portion could then be linked to sequence recognizing peptides that could lead to very high specificity. A restriction enzyme with such properties is FokI. Additionally FokI has the advantage of requiring dimerization to have nuclease activity and this means the specificity increases dramatically as each nuclease partner would recognize a unique DNA sequence. To enhance this effect, FokI nucleases have been engineered that can only function as heterodimers and have increased catalytic activity. The heterodimer functioning nucleases would avoid the possibility of unwanted homodimer activity and thus increase specificity of the DSB. Although the nuclease portion of both ZFNs and TALENs have similar properties, the difference between these engineered nucleases is in their DNA recognition peptide. ZFNs rely on Cys2-His2 zinc fingers and TALENs on TALEs. Both of these DNA recognizing peptide domains have the characteristic that they are naturally found in combinations in their proteins. Cys2-His2 Zinc fingers typically happen in repeats that are 3 bp apart and are found in diverse combinations in a variety of nucleic acid interacting proteins such as transcription factors. TALEs on the other hand are found in repeats with a one-to-one recognition ratio between the amino acids and the recognized nucleotide pairs. Because both zinc fingers and TALEs happen in repeated patterns, different combinations can be tried to create a wide variety of sequence specificities. Zinc fingers have been more established in these terms and approaches such as modular assembly (where Zinc fingers correlated with a triplet sequence are attached in a row to cover the required sequence), OPEN (low-stringency selection of peptide domains vs. triplet nucleotides followed by high-stringency selections of peptide combination vs. the final target in bacterial systems), and bacterial one-hybrid screening of zinc finger libraries among other methods have been used to make site specific nucleases.
Applications
Over the past decade, efficient genome editing has been developed for a wide range of experimental systems ranging from plants to animals, often beyond clinical interest, and the method holds a promising future in becoming a standard experimental strategy in research labs. The recent generation of rat, zebrafish, maize and tobacco ZFN-mediated mutants testifies to the significance of the methods and the list is expanding rapidly. Genome editing with engineered nucleases will likely contribute to many fields of life sciences from studying gene functions in plants and animals to gene therapy in humans. For instance, the field of synthetic biology which aims to engineer cells and organisms to perform novel functions, is likely to benefit from the ability of engineered nuclease to add or remove genomic elements and therefore create complex systems. In addition, gene functions can be studied using stem cells with engineered nucleases.
Listed below are some specific tasks this method can carry out:
- Targeted gene mutation
- Creating chromosome rearrangement
- Study gene function with stem cells
- Transgenic animals
- Endogenous gene labeling
- Targeted transgene addition
Targeted gene addition in plants
Genome editing using ZFN provides a new strategy for genetic manipulation in plants and is likely to assist engineering desired plant traits by modifying endogenous genes. For instance, site-specific gene addition in major crop species can be used for 'trait stacking' whereby several desired traits are physically linked to ensure their co-segregation during the breeding processes.Progress in such cases have been recently reported in Arabidopsis thaliana and Zea mays. In Arabidopsis thaliana, using ZFN-assisted gene targeting, two herbicide-resistant genes (tobacco acetolactate synthase SuRA and SuRB) were introduced to SuR loci with as high as 2% transformed cells with mutations. In Zea mays, disruption of the target locus was achieved by ZFN-induced DSBs and the resulting NHEJ. ZFN was also used to drive herbicide-tolerance gene expression cassette (PAT) into the targeted endogenous locus IPK1 in this case. Such genome modification observed in the regenerated plants has been shown to be inheritable and was transmitted to the next generation.
Several optimizations need to be made in order to improve editing plant genomes using ZFN-mediated targeting. These include the reliable design and subsequent test of the nucleases, the absence of toxicity of the nucleases, the appropriate choice of the plant tissue for targeting, the routes of introduction or induction of enzyme activity, the lack of off-target mutagenesis, and a reliable detection of mutated cases.
Gene therapy
The ideal gene therapy practice is that which replaces the defective gene with a normal allele at its natural location. This is advantageous over a virally delivered gene as there is no need to include the full coding sequences and regulatory sequences when only a small proportions of the gene needs to be altered as is often the case. The expression of the partially replaced genes is also more consistent with normal cell biology than full genes that are carried by viral vectors.
ZFN-induced targeting can also attack defective genes at their endogenous chromosomal locations. Examples include the treatment of X-linked severe combined immunodeficiency (X-SCID) by ex vivo gene correction with DNA carrying the interleukin-2 receptor common gamma chain (IL-2Rγ) with the correct sequence. Insertional mutagenesis by the retroviral vector genome induced leukemia in some patients, a problem predicted to be avoided by GEEN and ZFNs. However, ZFNs may also cause off-target mutations, in a different way from viral transductions. Currently many measures are taken to improve off-target detection and ensure safety before treatment.
Recently, Sangamo BioSciences (SGMO) introduced the Delta 32 mutation (a suppressor of CCR5 gene which is a co-receptor for HIV-1 entry into T cells therefore enabling HIV infection) using Zinc Finger Nuclease (ZFN). Their results were presented at the 51st Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) held in Chicago from September 17–20, 2011. Researchers at SGMO mutated CCR5 in CD4+ T cells and subsequently produced an HIV-resistant T-cell population.
Reference:
Genome editing with engineered nucleases. (2012, June 13). In Wikipedia, The Free Encyclopedia. Retrieved 10:59, July 23, 2012, from http://en.wikipedia.org/w/index.php?title=Genome_editing_with_engineered_nucleases&oldid=497435142







